
第一作者:陈赛、徐依依、常鑫(天津大学)通讯作者:巩金龙(天津大学)关键词:丙烷脱氢,金属-氧化物相互作用,缺陷态二氧化钛,包覆结构

相关研究成果以“Defective TiOx overlayers catalyze propane dehydrogenation promoted by base metals”为题发表在Science 期刊上,并被选为当期封面文章。
以下为转载X-MOL资讯公众号内容
全文速览
本工作基于对金属和氧化物相互作用本质的认知,提出利用氧化物与金属之间电子相互作用促进催化过程的研究思路。通过氢气还原控制氧化钛在金属镍颗粒表面的覆盖程度,实现了氧化钛和镍之间电子转移的精准调节。在丙烷脱氢反应条件下,该催化剂展现出与主流催化剂相当的活性、选择性和稳定性。机理研究表明,缺陷态氧化钛覆盖层中,氧空位邻近的四配位钛位点是丙烷脱氢的关键活性物种,次表面金属镍作为电子助剂,加速氧化钛表面碳氢键活化和氢气解吸过程,同时抑制裂解、积碳等副反应的发生,提升了丙烯收率。该制备策略提高了催化剂的活性和稳定性,实现了催化剂的循环使用,有望为丙烷脱氢制丙烯工艺提供新的解决方案,为推动化学工业的可持续发展提供新的思路和方法。
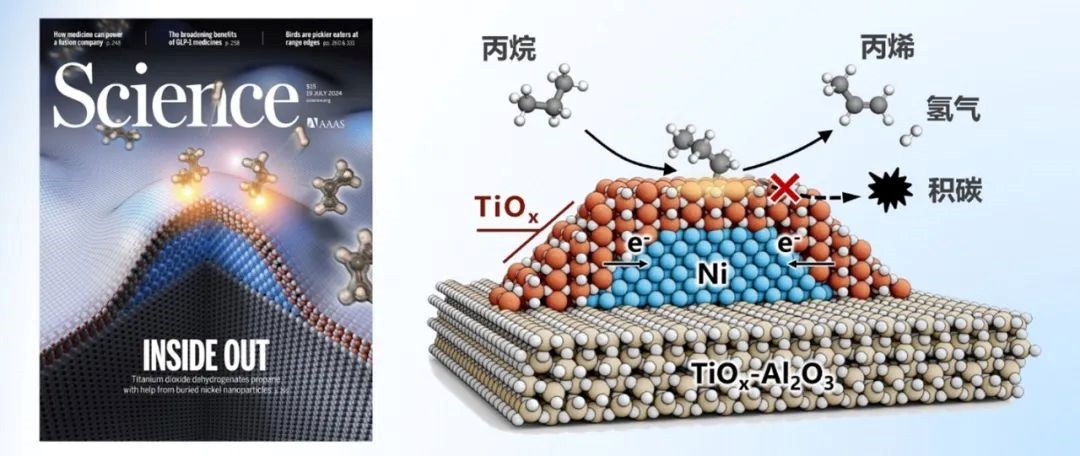
丙烯是重要的基础化工原料。在众多丙烯生产技术中,丙烷脱氢技术(PDH)因经济效益高、石油依赖低成为主流。Pt和CrOx基催化剂是两种主要的商业催化体系,但它们要么依赖于贵金属的使用,要么受限于自身毒性和积碳沉积导致的快速失活。在可持续发展要求下,工业界和学术界都在寻找成本低廉且对环境友好的廉价氧化物催化剂,包括VOx、GaOx、ZrO2和TiOx等。它们具有C–H键活化能力,但PDH本征活性较低。掺杂、气体预处理、晶面和晶相调控以及有机金属化学等在内的策略已被用于此类催化剂的设计和改性,但这些氧化物催化剂本征活性低的缺点依然有待解决。此外氧化物催化剂还会在PDH反应条件下因还原重构、烧结或积碳而失活。使用两种典型的 PDH 活性组分(如贵金属和氧化物)构建金属-氧化物复合催化剂是有效的解决方法之一,但贵金属的使用限制了潜在的工业应用。
研究目标
本论文提出了由两种环境友好但PDH本征活性较低的组分二氧化钛(TiO2)与金属镍(Ni)构建Ni@TiOx催化剂。在PDH反应条件下,该催化剂表现出可观的脱氢活性和选择性。高温(>550°C)氢气处理,实现TiOx覆盖层对Ni纳米颗粒(NP)的完全封装,且该结构在PDH反应条件下保持不变。原位表征、反应动力学与理论计算进一步证实,缺陷态TiOx包覆层中四配位Ti4C及周边氧空位(Ov)构成了PDH催化活性位点。亚表面金属Ni充当电子助剂,通过促进TiOx包覆层对C–H键活化并加速H2脱附,间接参与催化脱氢过程。
图文精读
原位观测TiOx覆盖层的形成
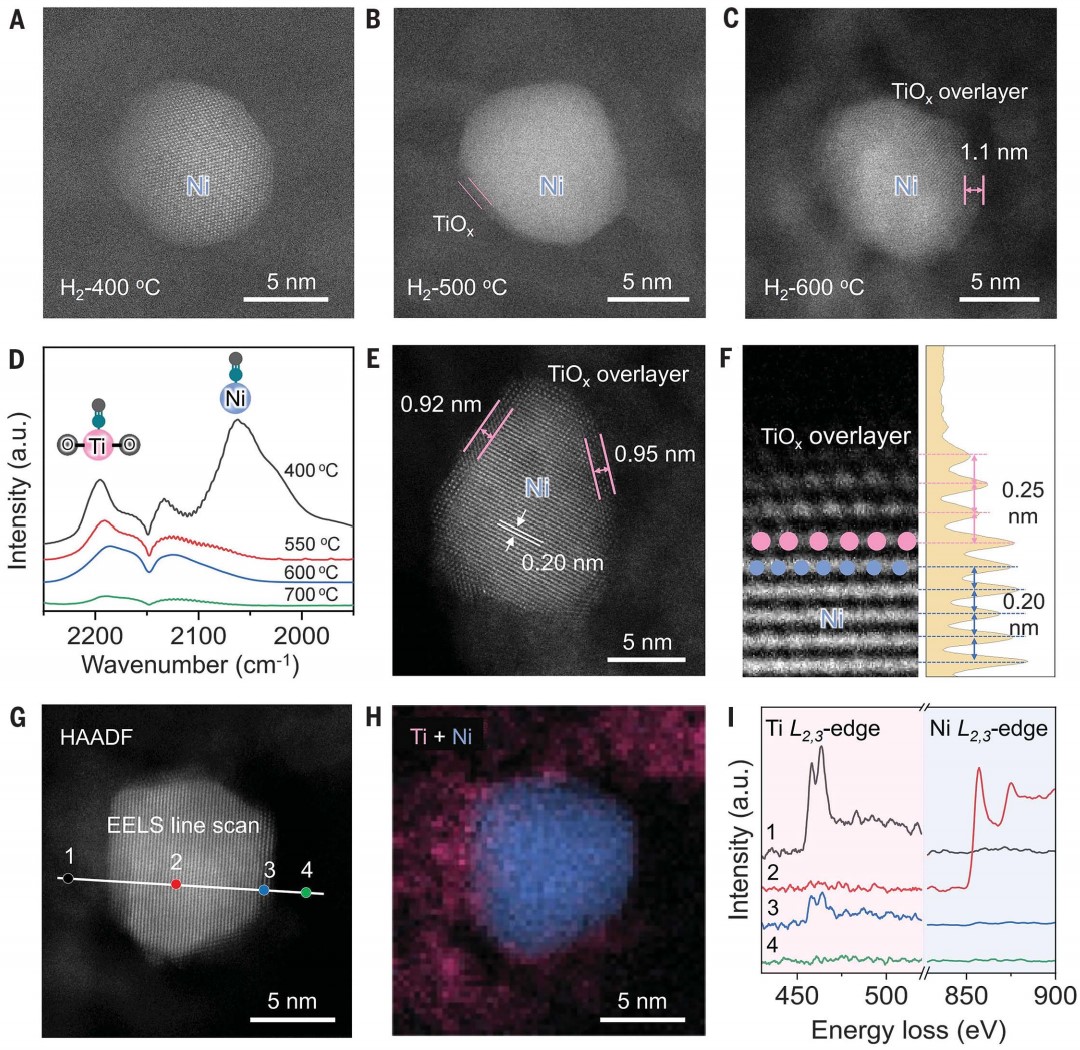
使用初湿浸渍法在TiOx/Al2O3纳米片上合成了一系列具有不同Ni负载量的Ni-Ti催化剂NixTiy/ Al2O3,其中 x 和 y 代表 Ni 与 Ti 的摩尔比。X 射线衍射 (XRD) 没有观察到属于任何结晶态 TiO2 的衍射峰,表明 TiOx 物种的高度分散。随着 Ni 负载量的减少,属于金属 Ni 的衍射峰逐渐消失,表明金属 Ni NP 尺寸减小,并且没有形成Ni-Ti合金。
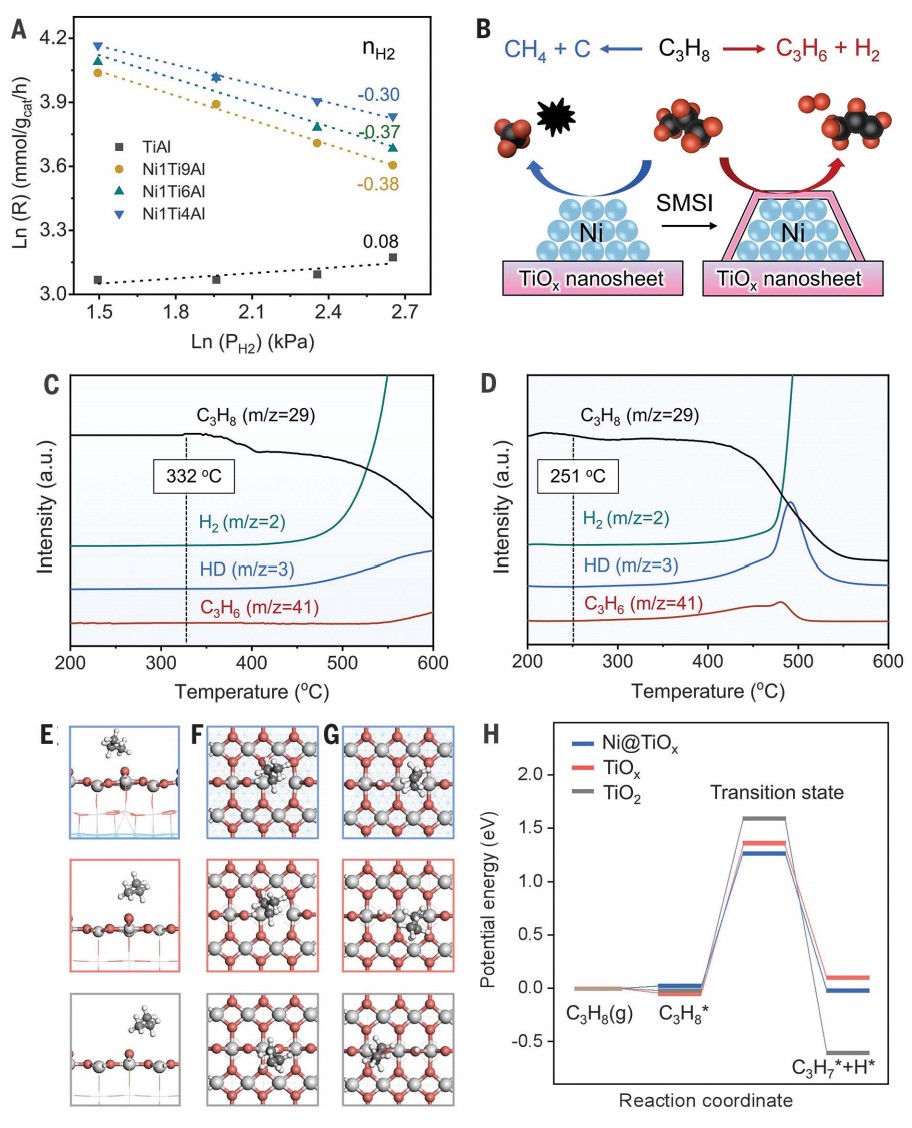
原位环境透射电子显微镜(ETEM)用于监测还原气氛下Ni NPs上TiOx覆盖层的演变。通过高角环形暗场扫描透射电子显微镜(HAADF-STEM)成像,确定在400 °C下 H2 还原 Ni1Ti4/Al2O3 形成了金属态 Ni NPs。当温度升高到 500°C(H2-500°C)时,在 Ni NPs 上探测到部分包覆的 TiOx 覆盖层。在 Ni@TiOx(H2-600°C)内形成了完全包覆且相对较厚(1 至 2 纳米)的 TiOx 覆盖层 (图1A-C),且在 600°C 退火后保持稳定,这与非原位观测到的核壳结构一致。包覆程度对还原温度的依赖性可以归因于高温下更强的金属-氧化物相互作用。漫反射红外傅里叶变换光谱 (DRIFTS) 观察到,在 400°C 氢气还原后,~2055 cm-1处出现了归属于金属 Ni 位点上的 CO 线性吸附的吸附峰,但随着还原温度升高到 600°C而消失(图1D),这说明金属Ni位点被TiOx包覆,这与原位 ETEM 观察到的包覆现象一致。Ni–Ni 和 Ti–Ti 的原子间距离分别为 0.20 和 0.25 nm,与晶格压缩的金红石 (110) 晶格间距一致,表明还原过程中 Ni 和 Ti 之间的相互作用导致了这种结构变化(图1E, F)。对于单个 Ni@TiOx NP中心和边缘点的电子能量损失谱 (EELS) 观测,揭示了Ti元素分布在不存在Ni元素的颗粒边缘(图1G-I)。表面敏感的低能离子散射光谱也证实了Ni@TiOx包覆结构的形成。
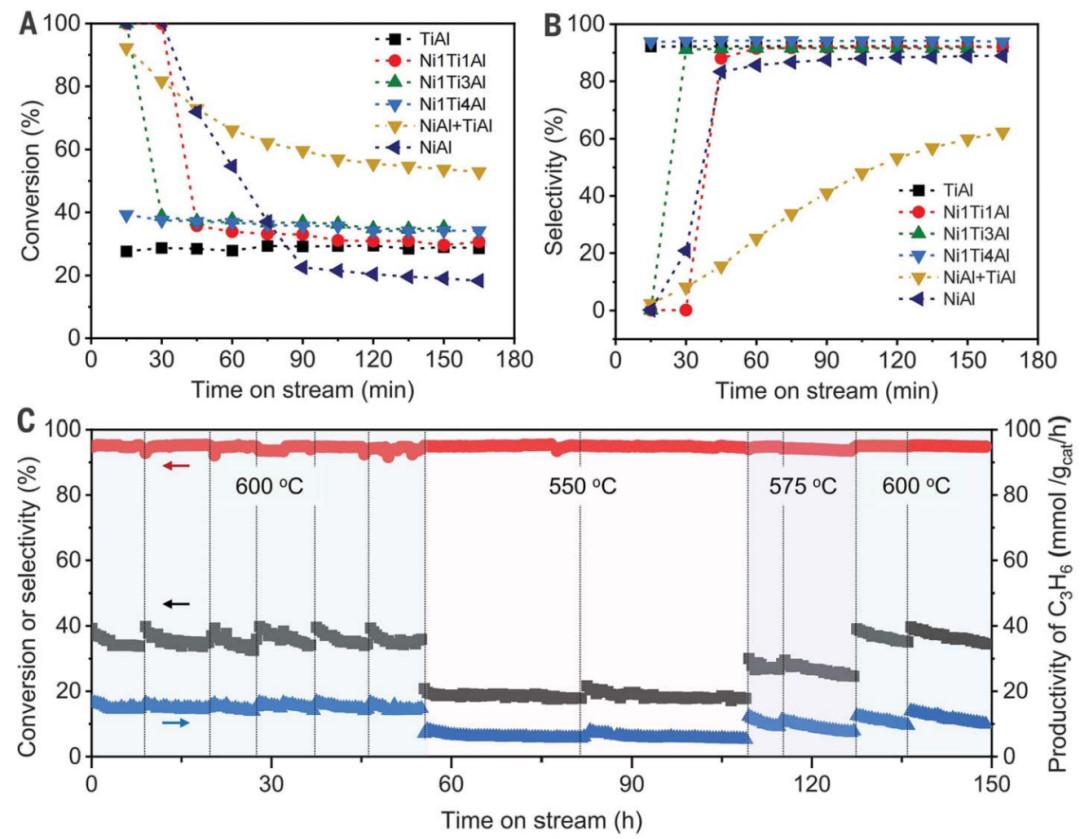
作者研究了 Ni@TiOx 催化剂的丙烷脱氢反应性能。由于金属Ni位点的C-C裂解活性高,Ni/Al2O3催化剂在600°C下对丙烯的初始选择性较差(0.4%),经历产甲烷并沉积焦炭的快速诱导期。而Ni1Ti1/Al2O3 和 Ni1Ti3/Al2O3 中金属Ni位点没有被不完全包覆,PDH反应初始阶段也观察到了类似的诱导期(图2A, B)。然而,对于Ni1Ti4/Al2O3(Ni@TiOx)催化剂,由于表面没有金属Ni位点暴露,生成甲烷的诱导期被消除,丙烯最高产率达到15.6 mmol gcat-1 h-1。而对于物理混合的Ni/Al2O3 和 TiOx/Al2O3,由于Ni 和 TiOx 之间的空间距离阻碍了金属-载体相互作用,导致PDH催化性能较差。相比之下,Ni@TiOx表现出优异的丙烯初始选择性,丙烯选择性接近94%,丙烷转化率接近40%,是商业锐钛矿型TiO2(7.8%)和金红型石TiO2(10.9%)的四倍左右。不同还原温度(550、575和600°C)形成的Ni@TiOx催化剂,在550°C性能测试中丙烯选择性和形成速率几乎一致。表明致密的TiOx覆盖层可以在高温(>550°C)下形成,并且在600°C下保持稳定,不会发生过度还原或重构,与CO-DRIFTS所观测结果一致。
在长程反应性能测试(图 2C)中,Ni@TiOx 催化剂对丙烯的选择性保持在~93%。丙烷转化率在共进料 H2情况下略有下降,但反应稳定性提升。PDH反应3 小时后热重分析(TGA)曲线显示Ni@TiOx的积碳量显著低于Ni/Al2O3。这表明TiOx覆盖层隔离了金属Ni与气相烃类的直接接触,从而有助于抑制PDH过程中Ni NPs上积碳的形成,这与O2程序升温氧化(TPO)和拉曼光谱对于积碳表征结果相对应。Ni@TiOx催化剂在550、575和600°C下的失活速率常数(kd)分别为0.007、0.018和0.073 h−1,远低于CrOx/Al2O3催化剂(600°C时为0.32 h−1);Ni@TiOx催化剂的性能优于大多数已报道的氧化物基催化剂,与贵金属 Pt 基催化剂相当。在连续脱氢再生循环中,初始活性可以完全恢复并保持稳定。再生后的Ni@TiOx催化剂几乎没有Ni和Ti元素的损失,XRD 和高分辨率透射电子显微镜图像也说明没有烧结或重构现象发生。
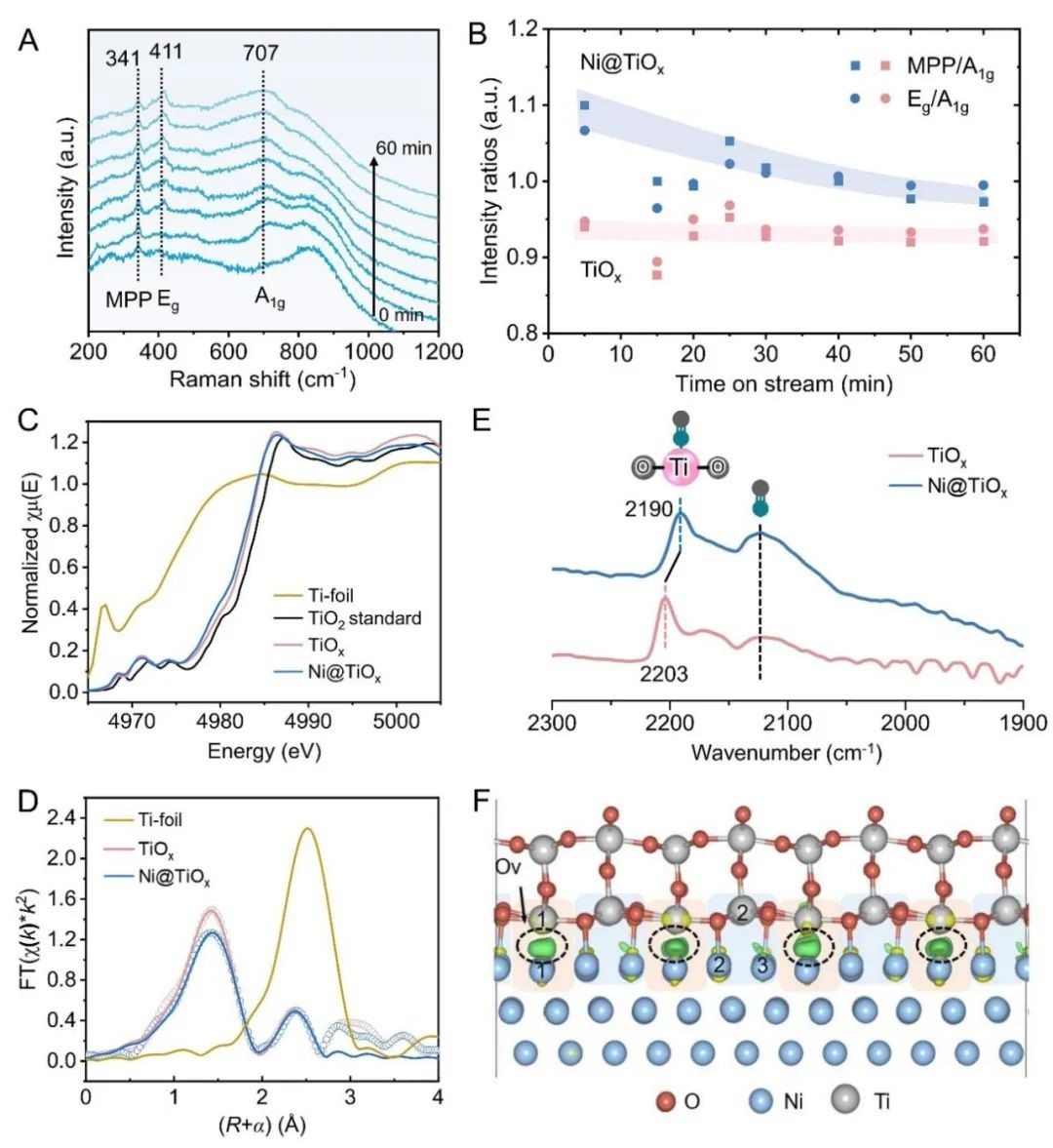
完全封装的Ni@TiOx催化剂具有更高的丙烷脱氢反应性能,表明活性位点可能位于TiOx 覆盖层上,而金属Ni间接参与催化过程。利用原位拉曼光谱研究TiOx的配位结构,在341和411 cm-1 处观察到对应于金红石型TiO2-Ti2O3 转变过程中中间O缺陷态的多光子过程(MPP)和平面O-O相互作用(Eg)模式的谱带(图3A)。在H2中暴露约60 分钟后,以707 cm−1 处的谱带(对应金红石型TiO2的O–Ti–O键(A1g)的对称伸缩振动)为参考,Ni@TiOx的MPP、Eg与A1g的强度比高于TiOx,这表明 Ni@TiOx的配位不饱和位点比例高于单纯的TiOx(图3B)。作者提出在H2还原过程中,H从金属Ni NPs 溢流到TiO2加速了界面处TiO2的还原,对应H2-程序升温还原(TPR)曲线中还原温度的下降和还原峰面积的增加。O 1s X射线光电子能谱(XPS)证实表面Ov浓度提高(27.5% vs. 19.8%),电子顺磁共振(EPR)观察到被捕获在Ov位点的未配对单电子信号增强。原位Ti 2p XPS显示Ni@TiOx 的 Ti3+/(Ti3++Ti4+)物种比例约为TiOx的两倍,表明TiOx覆盖层更容易被还原并形成配位不饱和Ti位点,且丙烯形成速率随Ti3+/(Ti3++Ti4+)比例的增加而增加。Ti K edge扩展X射线吸收精细结构(EXAFS)光谱的拟合结果显示,Ni@TiOx的Ti-O配位数为3.8,而 TiOx的Ti-O 配位数为4.2(图3C, D)。上述结果说明缺陷态TiOx覆盖层中四配位Ti4C位点与周边的氧空位可能作为活性更高的PDH反应位点。以CO的表面吸附为探针研究潜在的PDH反应活性位点,结果表明仅在2190 cm−1处观察到一个归属于四配位Ti4C的CO吸附带(图3E),而没有观察到Ni和Ni-TiOx界面上的CO吸附峰,表明Ni@TiOx催化剂表面的TiOx覆盖层作为活性位点,而不是金属Ni或Ni-TiOx界面位点。
为了从微观上解析催化机理,作者利用晶体外延生长的理念构建了金属镍被氧化钛全包覆(Ni@TiOx)的晶格匹配的理论模型,发现界面处含有Ov的Ni-TiOx相对于界面不含Ov的模型表现出更高的稳定性。Ni@TiOx的差分电荷密度计算表明界面失去电子,位于界面Ov位点的Ti1和Ni1原子获得电子,电子通过Ni-TiOx界面发生转移(图3F)。相比之下,位于非Ov位点的Ti2原子仅获得微量电子,同时Ni@TiOx表面Ti原子的净原子电荷与纯TiO2相当。值得注意的是,位于非Ov位点Ni2和Ni3原子的电荷变化幅度明显小于位于界面Ov位点的Ni1原子。这意味着界面Ov位点的存在使得外层氧化物和内层金属之间建立了更强的电子相互作用。富电子Ni位点(Niδ −)的形成也和Ni核中观察到的电子积累(单个模型中Ni核得到约6个电子)相一致。这种界面处的电子相互作用进一步反映到了表面上,导致表面O位点失去电子。理论计算表明,与TiO2(-0.49 eV)相比,Ni@TiOx表面O位点上H的平均吸附强度(0.04 eV)降低,说明在Ni@TiOx上H的脱附有利,印证了亚表面金属Ni和 TiOx覆盖层之间的电子相互作用改变了表面Ti-O位点的状态。实验上对600°C下D2预处理的Ni@TiOx催化剂进行H2-程序升温表面反应(TPSR),发现Ni@TiOx上活化的HD物种初始峰为98°C,而TiOx的初始峰为226°C,同样说明前者对H的吸附更弱。
相关催化剂研究
为了评估该设计策略在PDH反应中的有效性和普遍性,本论文研究了用Cu取代Ni或用ZnO取代TiOx的催化剂,观察到经高温氢气还原后,金属NPs被连续和均匀的氧化物覆盖层完全覆盖,丙烷转化率和丙烯选择性均高于相应的母体体系。600°C,Cu@TiOx催化剂获得了近36%丙烷转化率,近~94%丙烯选择性;NiZn@ZnO催化剂获得了近41.6%丙烷转化率,近~90%丙烯选择性,这些表明了金属颗粒表明的氧化物覆盖层对C–H键活化的有效性。
心得与展望
作者基于对金属和氧化物相互作用本质的认知,提出利用氧化物与金属之间的电子相互作用促进催化过程的科学假设,设计Ni@TiOx丙烷脱氢催化剂。我国是世界钛资源储量大国,镍资源也十分丰富。通过控制氧化钛在金属镍颗粒表面的覆盖程度,实现了氧化钛和镍之间电子转移的精准调节。实验和理论分析表明,氧空位邻近的四配位钛位点是丙烷脱氢的关键活性物种,次表面金属镍作为电子助剂,加速氧化钛表面碳氢键活化和氢气解吸过程,抑制裂解、积碳等副反应的发生。该制备策略提高了催化剂的活性和稳定性,实现了催化剂的循环使用,潜在降低了催化剂成本。该研究结果有望为丙烷脱氢制丙烯廉价环保催化剂提供新的解决方案,为推动化学工业的可持续发展提供新的思路和方法。
相关研究成果发表在Science 期刊上,并被选为当期封面文章。该研究得到国家重点研发计划、国家自然科学基金科学中心等项目的资助。

原文(扫描或长按二维码,识别后直达原文页面):Defective TiOx overlayers catalyze propane dehydrogenation promoted by base metalsSai Chen†, Yiyi Xu†, Xin Chang†, Yue Pan, Guodong Sun, Xianhui Wang, Donglong Fu, Chunlei Pei, Zhi-Jian Zhao, Dong Su, Jinlong Gong*Science, 2024, 385, 295-300, DOI: 10.1126/science.adp7379

